
December 3, 2008
La Jolla, CA — Long thought of as mere bystanders, astrocytes are crucial for the survival and well-being of motor neurons, which control voluntary muscle movements. In fact, defective astrocytes can lay waste to motor neurons and are the main suspects in the muscle-wasting disease amyotrophic lateral sclerosis (ALS).
To get to the root of this complicated relationship, researchers from the Salk Institute for Biological Studies for the very first time established a human embryonic stem cell (hESC)-based system for modeling ALS. Their study confirmed that dysfunctional human astrocytes turn against their charges and kill off healthy motor neurons. But more importantly, treating the cultured cells with apocynin, a powerful anti-oxidant, staved off motor neuron death caused by malfunctioning astrocytes.
Their findings, which appear in the Dec. 4 issue of the journal Cell Stem Cell, provide new insight into the toxic pathways that contribute to the demise of motor neurons in ALS and open up new possibilities for drug-screening experiments using human ALS in vitro models, as well as clinical interventions using astrocyte-based cell therapies.
"A variety of drugs that had demonstrated significant efficacy in mouse models didn't keep their promise in both preclinical and clinical trials," says Fred H. Gage, Ph.D., a professor in the Laboratory for Genetics, who led the study. In fact, just one drug—riluzole—has been approved by the FDA to treat ALS, and it only slows the course of the disease by two months.
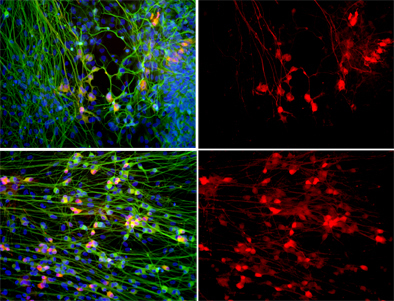
Top: When motor neurons (shown in red) are grown in the presence of defective astrocytes their numbers plummet. Bottom: Treating the cultures with apocynin, a powerful anti-oxidant, dramatically increases the survival of motor neurons. All cells' nuclei are labeled blue and neurons are shown in green. Right panel: Only motor neurons are shown.
Image: Courtesy of Dr. M. Carol Marchetto, Salk Institute for Biological Studies.
"There is an urgent need for new ALS models that have the potential to translate into clinical trials and that could, at a minimum, be used in conjunction with the murine models to verify drugs and drug targets," says Gage.
ALS, also known as Lou Gehrig's disease, was named after the legendary New York Yankee slugger who lent his name to the mysterious illness over 60 years ago. Usually fatal, the neurodegenerative disease attacks motor neurons controlling voluntary movement, leading to progressive paralysis and muscle atrophy.
Although ALS was first classified as a disease over 140 years ago, there are still few clues as to its cause. An important step toward understanding the disease came when scientists discovered that ALS can be induced by inherited mutations in the gene encoding the SOD1 enzyme, short for superoxide dismutase 1. This enzyme protects the body from damage caused by free radicals, highly reactive molecules produced by cells during normal metabolism.
Spinal motor neurons express high levels of SOD1, which many originally thought might explain their selective vulnerability. But soon, mouse experiments revealed that motor neuron degeneration is not necessarily associated with the expression of defective SOD1 in the motor neurons per se but rather with its expression in a critical number of neighboring support cells.
Since most treatments that worked in ALS mouse models didn't live up to expectations in preclinical and clinical trials, postdoctoral researcher and first author M. Carol Marchetto, Ph.D., looked for an alternative: "Transgenic mice containing the human mutated forms of SOD1 have been very useful to study the disease onset and progression. But we felt that cell culture models using both human neurons and astrocytes could potentially be very useful for drug screening and, to some extent, cell replacement therapies."
To uncover the contribution of astrocytes to human motor neuron degeneration, Marchetto first coaxed hESCs to differentiate into motor neurons through a series of physical manipulations and exposure to a number of growth factors. When she co-cultured these cells with human astrocytes expressing a mutated form of SOD1, the number of motor neurons alive in the Petri dish plummeted. "In the presence of the mutation, the astrocytes activated an inflammatory response and started producing reactive oxygen species, a hallmark of ALS," says Marchetto.
When she treated these cells with known antioxidants such as apocynin, which is found in many plants, epicatechin, one of the beneficial ingredients in green tea and chocolate, or alpha-lipoic acid, which is produced by the body, the percentage of astrocytes churning out harmful reactive oxygen species decreased significantly. Not only that, when she treated motor neurons cultured in the presence of mutant astrocytes, apocynin—the only one tested in a co-culture experiment—helped motor neurons withstand their no-longer-supportive environment.
"We believe that we can use this system as a rapid drug screening test for oxidative damage to identify the best candidates for subsequent long-term co-culture experiments," says Marchetto.
While research on the effects of the SOD1 gene mutation is providing important clues about the possible causes of motor neuron death, only a small fraction of all ALS cases are actually due to the mutation; other as yet unidentified genetic causes clearly exist.
"The rapid advances in induced pluripotent stem cell technology will soon allow us to generate patient-specific stem cells that can be used in our co-culture assay to gain new insight into the different causes of ALS," says Gage.
For information on the commercialization of this technology, please contact Mike White at 858-453-4100, x 1703 (mwhite@salk.edu) in of the Salk Office of Technology Management and Development.
This study was funded by Project ALS, the Dana and Christopher Reeve Foundation, the California Institute for Regenerative Medicine, the Lookout Fund, and the National Institutes of Health.
Researchers who also contributed to the work include postdoctoral researchers Alysson R. Muotri, Ph.D., and Yangling Mu, Ph.D., in the Gage laboratory, postdoctoral researcher Alan M. Smith, Ph.D., and assistant professor Gabriela G. Cezar, Ph.D., both at the University of Wisconsin-Madison, Madison.
Office of Communications
Tel: (858) 453-4100
press@salk.edu



