
March 11, 2012
Salk scientists' discovery explains how a class of chemotherapy drugs works
Salk scientists' discovery explains how a class of chemotherapy drugs works
LA JOLLA, CA—The well-being of living cells requires specialized squads of proteins that maintain order. Degraders chew up worn-out proteins, recyclers wrap up damaged organelles, and-most importantly-DNA repair crews restitch anything that resembles a broken chromosome. If repair is impossible, the crew foreman calls in executioners to annihilate a cell. As unsavory as this last bunch sounds, failure to summon them is one aspect of what makes a cancer cell a cancer cell.
A recent study from scientists at the Salk Institute for Biological Studies showed exactly how cells sense the possibility that their DNA is damaged as a first step to responding to the failure to divide properly. That study, published March 11 in Nature Structural and Molecular Biology, found that if cells take too long to undergo cell division, structures at the tips of their chromosomes, known as telomeres, send out a molecular SOS signal.

(R to L) Jan Karlseder, Professor, Molecular and Cell Biology Laboratory and Makoto Hayashi, research associate.
Image: Courtesy of the Salk Institute for Biological Studies
These findings have dual implications for cancer chemotherapy. First, they show how a class of anti-cancer drugs that slows cell division—known as mitotic inhibitors—kills cells. This class includes the common chemotherapy drugs Vinblastine, Taxol and Velcade. More significantly, the findings suggest ways to make therapy with those inhibitors more potent.
“How mitotic inhibitors work as cancer therapy has been a 25-year-old question,” says Jan Karlseder, a professor in Salk’s Molecular and Cell Biology Laboratory and the study’s senior author. “These drugs are widely used, but it was unclear why they actually kill cancer cells.”
The Karlseder lab has long been fascinated by the roles played by telomeres in aging and cancer. Often described as the genomic equivalent of the plastic caps that keep shoelaces from fraying, telomeres form a protective protein/DNA cap on each end of linear chromosomes, masking those ends from vigilant but apparently myopic repair proteins, who might mistake exposed chromosome ends for broken DNA.
Initially, the group searched for specific proteins that might keep telomeres intact during cell division. To do so, they genetically eliminated candidate proteins one by one and then examined cells using fluorescence microscopy to detect whether telomeres became damaged. What they found was unexpected—namely, that any manipulation that crippled and prolonged cell division produced increased numbers of punctate telomere blobs indicative of “unprotected” telomeres.
Treating cells with mitotic inhibitors used in cancer chemotherapy did the very same thing. Those studies established a link between arrested mitosis, telomere perturbation and cell death.
They then confirmed that as cells stalled in mitosis, telomeric shoelace caps started to disintegrate. “Normally the ends of chromosomes are covered by the protein TRF2, which protects the telomere,” says Makoto Hayashi, a postdoctoral fellow in the Karlseder lab and the study’s first author. “But we found that, during mitotic arrest TRF2 dissociates from telomeres, exposing chromosome ends as damaged DNA. That likely activates a DNA damage signal.”
The clincher came when the investigators found that the longer cells endured telomere distress, the more massively they induced what ought to be everyone’s most treasured gene, the tumor suppressor p53.
“DNA damage signals occurring during mitotic arrest likely predispose cells to upregulate p53 in the following phase,” says Hayashi. “This then either halts the cell cycle to enable DNA repair to occur or commits cells to a suicide pathway called ‘apoptosis’.”
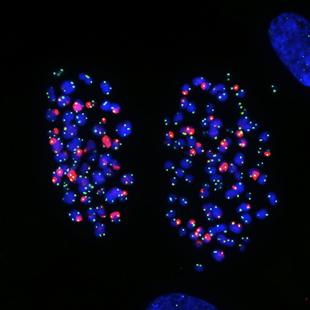
This microscope image shows chromosomes in human lung cells exhibiting telomere damage caused by colcemid, a drug that arrests cell division.
The Salk scientists discovered the molecular pathway that initiates a stress response upon treatment with chemotherapy drugs, resulting in a cessation of cell growth or cell death.
Image: Courtesy of Jan Karlseder, Professor, Molecular and Cellular Biology Laboratory, Makoto Hayashi, research associate and James Fitzpatrick, Waitt Advanced Biophotonics Center
Karlseder notes that p53 and proteins it mobilizes constitute the most frequently mutated pathway in cancer. “A functional p53 pathway is a healthy sign, as it is the cell’s first responder to DNA breaks or telomere dysfunction,” he says, “Without it, chromosomes could become unstable and could fuse to one another or break, leading to a loss of genome integrity and uncontrolled growth.”
This work suggests novel strategies that could be used in combinatorial cancer chemotherapy regimes, which rely on the synergy between two or more drugs. The theory is that a multi-pronged approach might pack more of a wallop than a sledgehammer alone, as evidenced by the highly effective “triple cocktail” of drugs now used to treat AIDS.
“To make therapy more effective and reduce side effects, we might be able to use more moderate levels of mitotic inhibitors, which at high doses can cause severe side effects, paired with a different drug that sensitizes cells to the DNA damage response,” says Karlseder. “That could improve the chances of catching 100 percent of the tumor cells.”
Prior to this study telomeres were already celebrated for the fact that they regulate aging by acting as a cellular clock, which ticks down a cell’s age. “Every time a cell divides, a little bit of the telomere cap is lost until it is gone, signaling that cells cannot divide anymore,” explains Karlseder. “This limitation ensures that cells do not become immortal or cancerous.”
Recent studies show, however, that among their other insidious activities, cancer cells figure out how to set back the telomere clock and keep them immortal. “They do this by switching on pathways that specifically lengthen telomeres,” says Karlseder. “These pathways therefore represent an important potential target for therapy.”
Also contributing to this study were Anthony Cesare, of the Karlseder Lab; James Fitzpatrick, director of Salk’s Waitt Advanced Biophotonics Core Facility; and Eros Lazzerini-Denchi, of The Scripps Research Institute in La Jolla.
Funding for the study came from the National Institutes of Health, the Human Frontier Science Program and the Japan Society for the Promotion of Science.
About the Salk Institute for Biological Studies:
The Salk Institute for Biological Studies is one of the world’s preeminent basic research institutions, where internationally renowned faculty probe fundamental life science questions in a unique, collaborative, and creative environment. Focused both on discovery and on mentoring future generations of researchers, Salk scientists make groundbreaking contributions to our understanding of cancer, aging, Alzheimer’s, diabetes and infectious diseases by studying neuroscience, genetics, cell and plant biology, and related disciplines.
Faculty achievements have been recognized with numerous honors, including Nobel Prizes and memberships in the National Academy of Sciences. Founded in 1960 by polio vaccine pioneer Jonas Salk, M.D., the Institute is an independent nonprofit organization and architectural landmark.
JOURNAL
Nature Structural and Molecular Biology
AUTHORS
Makoto T. Hayashi, Anthony J. Cesare, James A. J. Fitzpatrick, Eros Lazzerini-Denchi, and Jan Karlseder
Office of Communications
Tel: (858) 453-4100
press@salk.edu



