
January 29, 2009
La Jolla, CA–How are neurodegenerative diseases such as Alzheimer’s initiated, and why is age the major risk factor? A recent study of a protein called MOCA (Modifier of Cell Adhesion), carried out at the Salk Institute for Biological Studies, provides new clues to the answers of these fundamental questions.
Under normal circumstances, MOCA is a key member of the squadron charged with keeping Alzheimer’s disease at bay. A team of researchers led by Salk professor David Schubert, Ph.D., demonstrated what happens when MOCA goes on furlough. In the process Schubert identified a novel pathway with broad implications for both Alzheimer’s and other age-related neurodegenerative diseases.
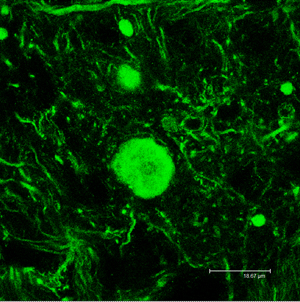
Over time, protein aggregates (shown in green) accumulate in the axons of MOCA-deficient mice. They start out small, initially causing few or no symptoms, but as they built up in the axons, they begin to destroy the cytoskeleton, the internal framework of the cells, increasingly interfering with the transmission of signals from the nerve cells. Eventually the affected axons die, followed by the death of the nerve cell itself.
Image: Courtesy of Dr. Qi Chen, Salk Institute for Biological Studies.
Their findings, reported in the current issue of the Journal of Neuroscience, show how neurodegenerative disease starts, initiating in the nerve ending and inducing gradual changes, like a chain reaction over a long time. The animal model used in the study also will allow scientists to better understand the processes behind the formation of the protein aggregates that are common to most neurodegenerative diseases. In addition, it will provide new opportunities to target the earliest steps for therapy.
MOCA was initially identified as a protein that binds to presenilin, a molecule that when mutated causes familial Alzheimer’s disease. MOCA is only found in neurons and regulates the expression of the beta amyloid protein responsible for the Alzheimer’s plaques that are the hallmark of the disease. To better understand MOCA’s function, Qi Chen, Ph.D., a senior scientist in Schubert’s laboratory, created a line of mice genetically engineered to lack the gene for MOCA.
“Because of the initial studies in cultured cells that we had done, we expected these mice to develop plaques,” explains Schubert. “What we found was that they develop ataxia–a motor coordination problem–as they age.” Chen then studied the pathology of these mice and found that it reflected a common feature of most age-related neurological diseases, not just Alzheimer’s.
The main problem turned out to be the degeneration of axons, the long projections that conduct impulses away from neurons. The axonal degeneration was caused by the accumulation of protein aggregates. Although the mice were not born with the problem, they acquired it, along with the ataxia, as they aged, and the ataxia worsened over time.
The aggregates started out small, initially causing few or no symptoms, but as they built up in the axons, they began to destroy the cytoskeleton, the internal framework of the cells, increasingly interfering with the transmission of signals from the nerve cells. Eventually the affected axons died, followed by the death of the nerve cell itself.
“Protein aggregates are common features of most age-related neurological diseases,” says Chen, the first author of the study. “So is axon degeneration; we see it in Alzheimer’s, ALS, and Huntington’s disease. Motor problems such as ataxia may be the most obvious manifestation because the aggregates appear in the long axons of the spinal cord. But the axonal aggregates also appear in the brain and may be the first step in the events that lead to age-associated neurological disease.”
After documenting the sequence of physiological and behavioral events that characterize the axon degeneration, Chen then sought to piece together the molecular pathway behind it, starting with MOCA and connecting findings from disparate studies that previously had identified parts of the pathway. He ended up with a single, step-by-step process for axon degeneration that for the first time linked together a number of diseases and conditions, including a form of mental retardation in humans.
“We had known that MOCA affected the cytoskeleton for some time, but no one had put together clear evidence showing how the sequential age-associated changes in the cytoskeleton of the nerve take place. Dr. Chen was able to do this, thereby connecting the disease pathology with the molecular biology,” says Schubert. “Now we know that MOCA is essential to the functional integrity of axons and have defined a complete pathway for axon degeneration.”
The study was funded with support from the National Institutes of Health, the Bundy Foundation, the Alzheimer’s Association, and the Shirley Foundation for Alzheimer’s Research.
In addition to Chen and Schubert, Paul E. Sawchenko, Ph.D., professor and laboratory head, and Charles A. Peto, both of the Salk Institute’s Laboratory of Neuronal Structure and Function, and Andrew Mizisin, Ph.D., professor, and G. Diane Shelton, Ph.D., adjunct professor, in the Department of Pathology at the University of California, San Diego, contributed to the study.
The Salk Institute for Biological Studies in La Jolla, California, is an independent nonprofit organization dedicated to fundamental discoveries in the life sciences, the improvement of human health, and the training of future generations of researchers. Jonas Salk, M.D., whose polio vaccine all but eradicated the crippling disease poliomyelitis in 1955, opened the Institute in 1965 with a gift of land from the City of San Diego and the financial support of the March of Dimes.
Office of Communications
Tel: (858) 453-4100
press@salk.edu



