Professor
Molecular and Cell Biology Laboratory
Director of the NCI-designated Salk Cancer Center
William R. Brody Chair

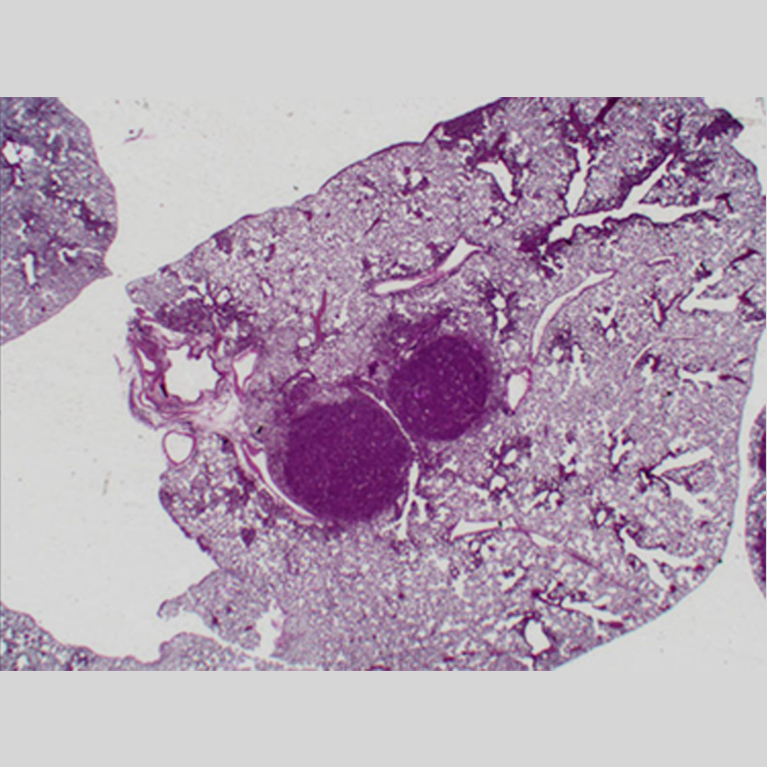
Lung cancer and type 2 diabetes are two leading causes of death in the United States. It turns out they have something else in common: both diseases involve a mishap in how cells use energy. In tumors, mutated cells usurp energy to grow aggressively. In diabetes, cells can no longer properly process and store two key sources of energy—sugar and fat (lipids).
Now, scientists have discovered a common set of biochemical pathways that normally suppress both cancer and type 2 diabetes. In the past decade there has been an explosion of interest in the details of how cancer pathways connect to metabolism, and, conversely, how metabolic pathways control development of cancer and diabetes. This has led to a number of new discoveries into how cells maintain their energy balance and couple their metabolism to their growth needs. Researchers are observing that multiple medications for diabetes and other metabolic disorders may help treat drug-resistant cancers and vice versa.
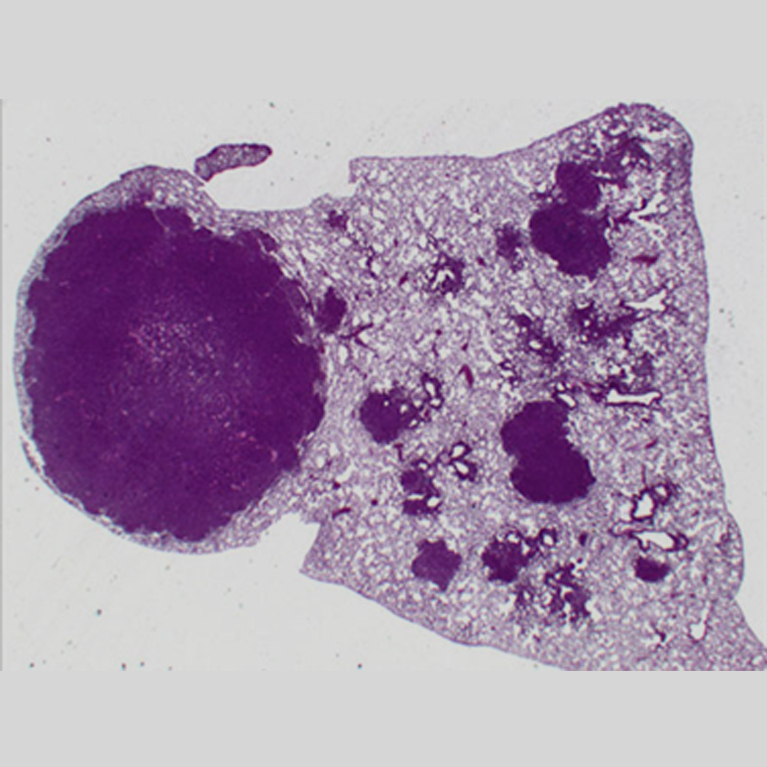
Fifteen years ago, Reuben Shaw discovered that a gene frequently mutated in cancer (LKB1) regulates an enzyme named AMPK. This enzyme is critical for the therapeutic benefit of metformin, which is currently the most widely used frontline type 2 diabetes medication. Ever since this discovery, Shaw wondered if drugs originally designed to treat metabolic diseases could also work against cancer.
This intriguing connection between cancer and metabolism coincided with emergent interest in metabolism in cancer, which rapidly moved to the forefront of cancer research. Shaw’s lab focuses on uncovering new aspects of a central metabolism and growth pathway that underpins how all cells respond to low nutrients and low energy. This AMPK pathway, which Shaw discovered, halts cell growth and reprograms metabolism when nutrients are scarce. The same pathway also helps to mechanistically connect the benefits of exercise, metformin and diet to the suppression of both cancer and diabetes. In the past decade at Salk, Shaw’s studies have led to the discovery of several new therapies for both cancer and metabolic diseases.
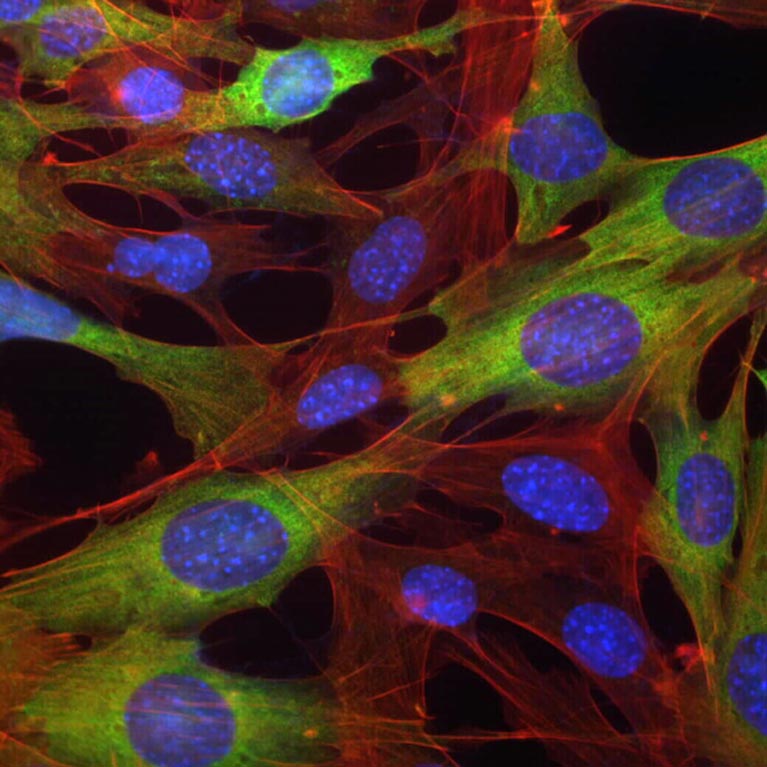
Shaw’s lab has discovered a way to target and stall fat synthesis to halt cancer growth. They developed the novel fat synthesis inhibitor drug called ND-646, which is promising when paired with common treatments for non-small-cell lung cancer. This discovery could lead to new therapeutic treatments for a variety of types of cancer such as liver or other lung cancers.
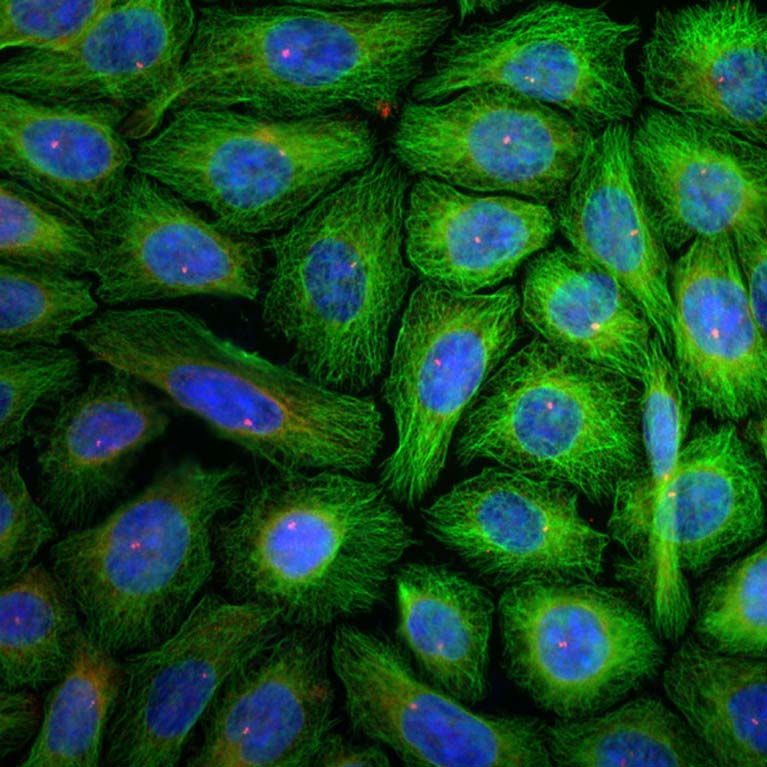
Shaw developed a new system to study how, where, and when AMPK carries out its molecular and therapeutic functions such as reversing diabetes, improving cardiovascular health, treating mitochondrial disease, and even extending life span. This novel model provides a new way to define the health benefits of AMPK, a master regulator of metabolism, for a variety of diseases.
Shaw’s lab discovered how cells trigger repair of their power generators, the mitochondria, following attacks. When cells are exposed to mitochondrial damage, the enzyme AMPK sends an emergency alert to the mitochondria instructing them to break apart into many tiny mitochondrial fragments to be reassembled into new, usable units. This finding provides insight for disorders such as Parkinson’s disease, which is linked to dysfunctional mitochondria.
BS Biology, Cornell University
PhD, Biology, MIT
Postdoctoral Fellow, Harvard Medical School