Fuente: https://www.illumina.com/
Secuenciación de lectura única frente a secuenciación de extremo emparejado
La secuenciación de lectura única implica secuenciar el ADN de un solo extremo y es la forma más sencilla de utilizar la secuenciación de Illumina. La secuenciación de extremos emparejados permite a los usuarios secuenciar ambos extremos de un fragmento y generar datos de secuencias alineables de alta calidad. La secuenciación de extremos emparejados facilita la detección de reordenamientos genómicos, como inserciones, deleciones o inversiones de genes y elementos de secuencia repetitivos, así como fusiones de genes y transcripciones novedosas. Dado que es más probable que las lecturas emparejadas se alineen con una referencia, la calidad de todo el conjunto de datos mejora.
Profundidad de secuenciación (cobertura)
La profundidad de cobertura es una medida del número de lecturas que se secuencian en un sitio genómico específico. La mayoría de los usuarios determinan el nivel de cobertura NGS necesario en función de la aplicación, así como de otros factores como el tamaño del genoma de referencia, el nivel de expresión génica, la literatura publicada y las mejores prácticas definidas por la comunidad científica.
- Para detectar mutaciones del genoma humano, SNP y reordenamientos, las publicaciones a menudo recomiendan una profundidad de cobertura de 10× a 30×, según la aplicación y el modelo estadístico.
- Para secuenciación de ARN, los investigadores suelen pensar en términos de millones de lecturas para muestrear. La detección de genes raramente expresados a menudo requiere un aumento en la profundidad de la cobertura.
- Para ChIP-Seq (secuenciación de inmunoprecipitación de cromatina), las publicaciones a menudo recomiendan una cobertura de alrededor de 100x.
¿Cuándo secuenciar más?
En los experimentos de secuenciación de Illumina, es muy fácil aumentar la nanocobertura o la profundidad de la secuencia, si luego decide que necesita más datos. Puede secuenciar más y combinar la salida de secuenciación de diferentes celdas de flujo. Hay una serie de razones para secuenciar más de la cobertura estimada originalmente, estas incluyen:
- Los efectos que ve no son estadísticamente significativos. La secuenciación de más lecturas generalmente aumentará la potencia de su ensayo.
- Estás investigando eventos que son muy raros. Por ejemplo, es posible que desee ver las transcripciones que se expresan a un nivel muy bajo en la secuenciación de ARN o las actividades de unión muy bajas en la secuenciación de ChIP.
- Ciertas revistas o campos pueden requerir un mayor nivel de cobertura para su aplicación en particular.
- Ciertos genomas pueden necesitar más secuenciación. Por ejemplo, ciertas regiones pueden ser difíciles de secuenciar y requieren más cobertura, o el genoma puede ser poliploide.
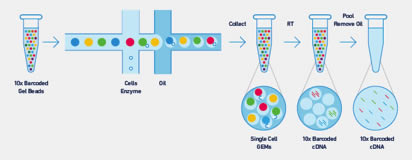
 El Sistema Sequel se basa en nuestro probado
El Sistema Sequel se basa en nuestro probado 
