
17 de noviembre.
LA JOLLA, CA–Con pocas excepciones, los genes saltadores, fragmentos inquietos de ADN que pueden moverse libremente por el genoma, se ven obligados a quedarse quietos. Sin embargo, en pacientes con síndrome de Rett, una mutación en el gen MeCP2 moviliza los llamados retrotransposones L1 en las células cerebrales, reorganizando sus genomas y posiblemente contribuyendo a los síntomas de la enfermedad cuando encuentran su camino hacia los genes activos, informan investigadores del Salk. Instituto de Estudios Biológicos.
Sus hallazgos, publicados en la edición del 18 de noviembre de 2010 de la revista Nature, no solo podría explicar cómo una sola mutación puede causar la desconcertante variabilidad de los síntomas típicos del síndrome de Rett, sino que también arrojaría nueva luz sobre la complejidad de los eventos moleculares que subyacen a los trastornos psiquiátricos como el autismo y la esquizofrenia.
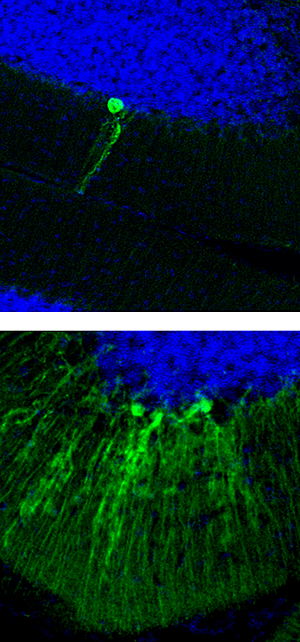
Los retrotransposones L1 son particularmente activos en el cerebelo, que juega un papel importante en el control motor. Las celdas verdes representan neuronas con nuevas inserciones L1
Imagen: Cortesía de la Dra. Carol Marchetto, Instituto Salk de Estudios Biológicos.
"Esta es la primera vez que podemos mostrar una conexión entre la estabilidad genómica y un trastorno mental", dice el autor principal. fred gage, Ph.D., profesor en el Laboratorio de Genética de Salk y titular de la Cátedra Vi and John Adler para la Investigación de Enfermedades Neurodegenerativas Relacionadas con la Edad. “En general, el mosaicismo genómico, generado por la retrotransposición L1, probablemente requiera una regulación estricta. Los mecanismos epigenéticos como la metilación son ideales para controlar la actividad de L1”.
"Ciertamente hay un componente genético en el síndrome de Rett y otros trastornos psiquiátricos, pero puede que no sea lo único relevante", dice la primera autora Alysson Muotri, Ph.D., quien comenzó el estudio como investigadora postdoctoral en el laboratorio de Gage y ahora ocupa un puesto como profesor asistente en el Departamento de Pediatría/Medicina Celular y Molecular de la Facultad de Medicina de la Universidad de California en San Diego. “Las inserciones y alteraciones somáticas causadas por elementos L1 podrían desempeñar un papel significativo pero subestimado porque son difíciles de detectar”, agrega.
El síndrome de Rett, una rara enfermedad del neurodesarrollo, afecta principalmente a niñas y se considera uno de los trastornos del espectro autista. La mayoría de los bebés parecen crecer y desarrollarse normalmente al principio, pero con el tiempo, los niños con síndrome de Rett tienen problemas cada vez mayores con el movimiento, la coordinación y la comunicación.
Las características típicas incluyen pérdida del habla, movimientos estereotípicos, retraso mental y problemas de comportamiento social. Aunque casi todos los casos son causados por una mutación en el gen MeCP2, la gravedad de los síntomas del síndrome de Rett afecta a las personas de forma muy variable.
Gage y su equipo encontraron la sorprendente conexión entre el síndrome de Rett y los retrotransposones L1 demasiado activos, cuando preguntaron cómo se regula su actividad en el cerebro. En trabajos anteriores, ya habían demostrado que los retrotransposones L1, a través de un mecanismo de "copiar y pegar", agregan cientos de copias adicionales al genoma de las neuronas, cambiando aleatoriamente la información en células cerebrales individuales. Sin embargo, solo estuvieron activos durante un breve período de tiempo: las primeras etapas del desarrollo de las células nerviosas. Una vez que las células precursoras neuronales se comprometieron con un destino neuronal, los elementos L1 fueron nuevamente abandonados en su lugar.
"Queríamos saber cómo se controla este proceso en el cerebro y qué sucede cuando el proceso sale mal", explica la investigadora posdoctoral y coautora Carol Marchetto, Ph.D. Estaban especialmente interesados en MeCP2, que desempeña un papel en la regulación de la actividad génica y que habían encontrado en la región reguladora de los elementos LINE-1.
Los experimentos iniciales encontraron que MeCP2 interfiere con la capacidad de un elemento L1 artificial para insertar copias adicionales de sí mismo en el genoma de las células madre neurales cultivadas. El elemento artificial había sido diseñado genéticamente para hacer que las células brillaran de color verde después de haberse extendido a un nuevo lugar.
Sin MeCP2 para mantenerlos en silencio, el retrotransposón L1 es libre de moverse por el genoma de las neuronas. Los puntos verdes representan células cerebrales con nuevas inserciones L1. El bulbo olfatorio se muestra en rojo, el cuerpo estriado en magenta y el cerebelo en cian.
Vídeo: Cortesía de los Dres. Carol Marchetto y Alysson Muotri con permiso de Nature
Cuando los investigadores de Salk rastrearon el mismo elemento indicador en los cerebros de ratones que carecían del gen para MeCP2, su actividad aumentó hasta seis veces en comparación con los ratones normales (ver película). "El aumento de la actividad fue específico de ciertas regiones del cerebro", dice Marchetto, "y se correlacionó con las regiones del cerebro que muestran diferencias fisiológicas cuando se elimina el gen MeCP2 en ratones".
Intrigados, querían saber si el síndrome de Rett, causado por mutaciones en el gen MeCP, conduce a una mayor movilidad de los transposones L1. Marchetto generó células madre pluripotentes inducidas (iPS) a partir de fibroblastos de un paciente de Rett y de un individuo no afectado, introdujo el elemento indicador y las diferenció en células precursoras neuronales. “La movilidad en las células derivadas del paciente de Rett era casi el doble en comparación con las células normales”, dice Marchetto.
Para confirmar que no solo su elemento L1 artificial estaba en movimiento, sino también elementos endógenos, Muotri y sus colegas desarrollaron una nueva técnica que les permitió detectar el aumento sutil en el contenido de ADN causado por copias adicionales de L1 insertadas en el genoma. Usando este método, pudieron demostrar que la cantidad de elementos L1 en los cerebros de los pacientes con RTT era significativamente mayor que en los cerebros de los controles.
"Las altas tasas de transposiciones neuronales en ratones con MeCP2 knock-out y pacientes con Rett pueden ser una consecuencia, más que una causa, del proceso de la enfermedad", dice Gage. "Sin embargo, las nuevas inserciones somáticas, especialmente durante las primeras etapas de desarrollo, pueden desempeñar un papel en las etapas posteriores de la enfermedad y podrían explicar la amplia variabilidad de los síntomas observados en los pacientes con síndrome de Rett".
Los investigadores que también contribuyeron al trabajo incluyen a Nicole G. Coufal y Ruth Oefner en el Laboratorio de Genética del Instituto Salk, Gene Yeo en el Departamento de Medicina Celular y Molecular de la Universidad de California, San Diego, así como a Kinichi Nakashima en el Laboratorio de Neurociencia Molecular del Instituto de Ciencia y Tecnología de Nara, Japón.
El trabajo fue financiado en parte por los Institutos Nacionales de Salud, la Fundación Emerald, la Fundación Mathers y el Fondo Lookout.
Sobre el Instituto Salk de Estudios Biológicos:
El Instituto Salk de Estudios Biológicos es una de las instituciones de investigación básica más importantes del mundo, donde profesores de renombre internacional investigan cuestiones fundamentales de las ciencias de la vida en un entorno único, colaborativo y creativo. Centrados tanto en el descubrimiento como en la orientación de futuras generaciones de investigadores, los científicos de Salk realizan contribuciones innovadoras a nuestra comprensión del cáncer, el envejecimiento, el Alzheimer, la diabetes y las enfermedades infecciosas mediante el estudio de la neurociencia, la genética, la biología celular y vegetal y disciplinas relacionadas.
Los logros de la facultad han sido reconocidos con numerosos honores, incluidos premios Nobel y membresías en la Academia Nacional de Ciencias. Fundado en 1960 por el pionero de la vacuna contra la polio Jonas Salk, MD, el Instituto es una organización independiente sin fines de lucro y un hito arquitectónico.
El Instituto Salk celebra con orgullo cinco décadas de excelencia científica en investigación básica.
Oficina de Comunicaciones
Tel: (858) 453-4100
prensa@salk.edu