Technologies Available for Collaboration
Neuroscience
Neurological disease modeling – Fred Gage Details»
Fred “Rusty” Gage is a world leader in neurogenesis and president of the International Society for Stem Cell Research. He has developed novel cellular models of neurological diseases (schizophrenia, MS, autism, Alzheimer’s, Parkinson’s) using human induced pluripotent stem cells (iPSCs) derived from donor patients and differentiated to specific neuronal cell types. These models are useful for studying disease mechanisms, identifying targets, screening compounds and stratifying patients. They are more predictive of disease phenotypes than current models. Gage has also developed an in vitro myelination model using iPSCs to be used for screening of pharmacological compounds and to better understand diseases such as schizophrenia and Parkinson’s.
Animal models of neuronal loss in diseases such as Parkinson’s – Martyn Goulding Details»
The fundamental mechanisms that contribute to the cognitive and motor deficits that accompany neurodegenerative diseases are poorly understood, due in large part to the progressive nature of these diseases. Goulding’s lab studies the immediate and downstream effects of neuronal cell loss, such as ongoing changes to spared neurons or to other cell types that make up the circuit, and determines how they adapt to the loss of a particular neuronal cell type. By selectively ablating specific neuronal cell types, in a time- and spatially-dependent manner, one can perform “surgical” lesions and examine the resultant changes with much greater precision. This allows for a more precise evaluation of the attendant cellular, synaptic and physiological changes that occur in response to the initial lesion. He has used this approach to develop a model of Parkinson’s disease.
Novel molecular targets for treating schizophrenia – Kuo-Fen Lee Details»
Dysfunctions in the glutamate, GABA and serotonin systems have also been implicated in schizophrenia. Most mechanistic studies have so far been focused on DA modulation of the neural circuitry in the prefrontal cortex (PFC) or the striatum, and related them to behavioral phenotypes of schizophrenia. However, little is known about the upstream regulatory circuits of these pathways. To best understand how cholinergic neurons exert their functions in distinct pathways and how to overcome the difficulty of mAChR subtype selectivity and peripheral side effects, Lee’s group has profiled and characterized genes that are uniquely or selectively expressed in cholinergic neuron subtypes and which uniquely regulate electrophysiology of mAChR subtypes. His lab is currently investigating the organization and function of a vastly underexplored but potentially highly important neural pathway from the cholinergic MT to DA neurons, and seeking potential causes, as well as molecular and cellular targets for treating schizophrenia.
Identifying Astrocyte-Secreted Factors to Treat Autism Spectrum Disorders – Nicola Allen Details»
Background and Significance
Multiple types of autism spectrum disorders (ASD) give rise to similar cellular defects in the developing brain – this is despite ASD being caused by distinct sets of genetic mutations or arising sporadically. Cellular phenotypes include altered neuronal death, reduced neuronal dendritic arbor growth, and defects in neuronal synapse formation and function. The similar cellular phenotypes present across multiple ASDs suggests they may share common defects in the molecular mechanisms that regulate neuronal development, and raises the potential of identifying common pathways for therapeutic targeting and treatment of sporadic ASD. Many features of ASD are non-cell autonomous for neurons, and can be induced by astrocytes, the most abundant cell type in the brain. Astrocytes from ASD mouse models induce dysfunction in wild type (WT) neurons, and WT astrocytes can rescue much of the dysfunction of ASD neurons (Fig 1). Astrocytes have important roles in brain development by secreting factors that regulate neuronal survival, dendrite outgrowth, synapse formation, and neuronal network function. Our lab has extensive experience in identifying and characterizing astrocyte-secreted factors in normal brain development, and we are applying this knowledge to identify core alterations in astrocyte secretion responsible for defects found in multiple types of ASD.
Hypothesis
We hypothesize that multiple forms of ASD share common defects in astrocyte-secretion of neuron-supportive factors, and that these defects impair neuronal maturation and comprise a novel target for therapeutic intervention in multiple forms of ASD.
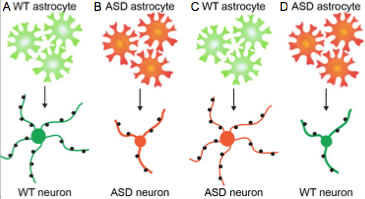 Figure 1. Astrocytes contribute to the pathology of ASD. A. WT astrocytes (green) secrete factors that promote the development of healthy neurons (green): neuron has a large central cell body, extends many long thin dendritic processes, and receives numerous syaptic connections (black). B. When ASD astrocytes (red) are cultured with ASD neurons (red) neuronal development is impaired; the neuron has a smaller cell body, extends less dendritic processes which are thick and stunted, and receives less synaptic connections. C. WT astrocytes release factors that are sufficient to rescue the phenotype of ASD neurons. D. ASD astrocytes release factors that are sufficient to impair the development of WT neurons.
To test this hypothesis we have:
1) Developed a protocol to purify and culture astrocytes from the postnatal mouse brain, in serum-free conditions to prevent induction of inflammatory genes.
2) Determined that proteins secreted by these astrocytes are sufficient to maintain neuronal survival and process outgrowth in vitro
3) Used mass spectrometry to identify the full complement of astrocyte-secreted proteins, a list of ~2000
4) Used RNA sequencing to identify astrocyte-expressed genes, and whether gene expression correlates with protein level.
We are now applying these methods to astrocytes from multiple models of ASD, to identify differentially secreted proteins that are candidates for the negative impact of ASD astrocytes on WT neurons. These candidates will be tested for their ability to regulate neuronal survival, neuronal process outgrowth, and synapse formation using in vitro assays.
This analysis of astrocyte-secretion, both in health and in ASD, will provide the first comprehensive analysis of the astrocyte secretome and identify novel targets for future therapeutic targeting in ASD.
Figure 1. Astrocytes contribute to the pathology of ASD. A. WT astrocytes (green) secrete factors that promote the development of healthy neurons (green): neuron has a large central cell body, extends many long thin dendritic processes, and receives numerous syaptic connections (black). B. When ASD astrocytes (red) are cultured with ASD neurons (red) neuronal development is impaired; the neuron has a smaller cell body, extends less dendritic processes which are thick and stunted, and receives less synaptic connections. C. WT astrocytes release factors that are sufficient to rescue the phenotype of ASD neurons. D. ASD astrocytes release factors that are sufficient to impair the development of WT neurons.
To test this hypothesis we have:
1) Developed a protocol to purify and culture astrocytes from the postnatal mouse brain, in serum-free conditions to prevent induction of inflammatory genes.
2) Determined that proteins secreted by these astrocytes are sufficient to maintain neuronal survival and process outgrowth in vitro
3) Used mass spectrometry to identify the full complement of astrocyte-secreted proteins, a list of ~2000
4) Used RNA sequencing to identify astrocyte-expressed genes, and whether gene expression correlates with protein level.
We are now applying these methods to astrocytes from multiple models of ASD, to identify differentially secreted proteins that are candidates for the negative impact of ASD astrocytes on WT neurons. These candidates will be tested for their ability to regulate neuronal survival, neuronal process outgrowth, and synapse formation using in vitro assays.
This analysis of astrocyte-secretion, both in health and in ASD, will provide the first comprehensive analysis of the astrocyte secretome and identify novel targets for future therapeutic targeting in ASD.
Figure 1. Astrocytes contribute to the pathology of ASD. A. WT astrocytes (green) secrete factors that promote the development of healthy neurons (green): neuron has a large central cell body, extends many long thin dendritic processes, and receives numerous syaptic connections (black). B. When ASD astrocytes (red) are cultured with ASD neurons (red) neuronal development is impaired; the neuron has a smaller cell body, extends less dendritic processes which are thick and stunted, and receives less synaptic connections. C. WT astrocytes release factors that are sufficient to rescue the phenotype of ASD neurons. D. ASD astrocytes release factors that are sufficient to impair the development of WT neurons.
Repair of synaptic connections following injuries such as stroke – Nicola Allen Details»
Allen’s lab investigates the molecular pathways that lead to connections between neurons, known as synapses, in the developing brain. Her group focuses on signaling interactions between neurons and astrocytes, a class of star-shaped glial cell. The hypothesis is that astrocytes play a crucial role in dictating synapse formation and function via the release of specific proteins that determine the type of synapse that will form, and the strength of that synapse. They are exploring whether these developmental findings can be used to address diseases such as stroke, by promoting the repair of synaptic connections following injury.
Astrocyte Regulation of Neuronal Synapses and Circuits – Nicola Allen Details»
Synapses are essential points of information transfer within neuronal circuits, and the correct formation and maturation of synapses is necessary for the brain to function throughout life. Astrocytes are the most abundant cell type in the brain, and many synapses have an astrocyte process associated with them. Landmark studies in the developing brain have shown that astrocytes secrete signals that can induce the formation of new synapses, alter the strength of synapses or cause the elimination of existing synapses. However the identity of all of these signals, and their specific mode of action, are not known. Our lab is working to identify how astrocytes interact with and regulate neuronal synapses throughout life: in development of neuronal circuits, in adult synaptic plasticity, and in synapse loss in aging.
</p
Specific areas of research:
- Identification and characterization of astrocyte-secreted signals that regulate neuronal synapse number and strength during development
- Determining what controls the release of synaptogenic factors from astrocytes
- Identifying how alterations in astrocyte protein secretion contribute to autism spectrum disorder
- Investigating if astrocytes in the adult brain limit synaptic plasticity and inhibit learning
- Investigating if astrocytes in the aging brain contribute to age-related synapse loss
- Determining if secreted signals from young astrocytes can be used to repair synapses after injury
- In vitro cultures of purified neurons and purified astrocytes, isolated using immunopanning, to identify novel astrocyte-secreted synaptogenic molecules and to dissect out the signaling pathways they regulate in neurons
- RNAseq and mass spectrometry to identify the astrocyte-specific transcriptome and secretome, across development both in vitro and in vivo
- Astrocyte-specific knock out mouse models to determine how identified factors regulate synapse and circuit formation and function in vivo
- Approaches such as electrophysiology to assess synaptic strength and plasticity, immunohistochemistry to assess synapse number, and behavioral assays to assess learning
Optogenetics in the treatment of Parkinson’s disease – Xin Jin Details»
Numerous motor and mental disorders, from Parkinson’s disease to obsessive-compulsive disorder, have been linked to the dysfunction of basal ganglia circuits. Jin’s lab employs a vast array of tools, including quantitative behavior, genetics and optogenetics, in vivo physiological and optical techniques to dissect the neural circuits and molecular mechanisms underlying action learning and selection in freely behaving mice.
Imaging of specific neuronal cell types in awake, behaving animals – Axel Nimmerjhan Details»
>Glial cells, the second major cell type in the brain, account for about ninety percent of human brain cells and more than fifty percent of the brain’s volume. Over the past few years, it has become clear that glial cells make crucial contributions to the formation, operation and adaptation of neural circuitry. Work in Nimmerjahn’s lab is centered on innovating light microscopic tools that enable the study of these electrically largely non-excitable cells and their interaction with other cells in the intact mammalian brain. They have created tools for cell-type-specific staining and genetic manipulation, for imaging cellular dynamics in awake, behaving mammals and for automated analysis of large-scale imaging data. This work has broad implications for our view of glial cells, the way information is processed in the brain, the interpretation of functional brain imaging signals and the treatment of neurodegenerative brain disease.
In vitro model of myelination and MS – Fred Gage Details»
Fred “Rusty” Gage is a world leader in neurogenesis and president of the International Society for Stem Cell Research. He has developed novel cellular models of neurological diseases (MS, autism, Alzheimer’s, Parkinson’s) using human induced pluripotent stem cells (iPSCs) derived from donor patients and differentiated to specific neuronal cell types. He has shown these to be useful for studying disease mechanisms, identifying targets and screening test compounds, and to be more predictive of disease phenotypes than current models. Dr. Gage has also developed an in vitro myelination model using iPSCs to be used for screening of pharmacological compounds and to better understand diseases such as MS.
Summary of myelination model:
Myelin is an electrically insulating material that forms a membrane layer, the myelin sheath, around the axon of a neuron. It is produced by oligodendrocytes in the central nervous system and Schwann cells in the peripheral nervous system. Myelination increases speed of impulse propagation by saltatory conduction through uninsulated portions of axon called Nodes of Ranvier. Loss of myelin membrane, as occurs in many neurological disorders such as multiple sclerosis, leads to disruption of electrical communication between neurons and permanent neuronal functional deficits including loss of motor control and cognition impairment. Multiple sclerosis alone affects over 400,000 people in the United States and more than 2.1 million people worldwide.
The myelination process has mostly been studied with isolated primary cells, in postmortem tissues, or with animal models. All approaches are restricted by variability among individuals, by the intrinsic differences between animal and human models and by the limited number of cells and animals, making large-scale experiments difficult. Embryonic stem cells (ESCs) allow for a relatively uniform population of a large number of cells and induced pluripotent stem cells (iPSCs) offer an opportunity to compare healthy and patient cells directly. We have established an in vitro (in a dish) myelination model using ESC- and iPSC-derived oligodendrocytes and neurons. Our assay will contribute to basic understanding of myelin formation leading to refined treatment strategies. Due to the reproducibility and reliability, our proposed assay will be available for pharmacological compounds screening and adaptation by other research groups to test their hypotheses. Currently, we achieved successful myelin formation using mouse ESCs and we are already gaining knowledge on the mechanics of myelin wrapping.
Oncology
A cell-based screen to identify therapeutic compounds for pancreatic cancer – Ronald Evans Details»Desmoplastic stroma is a defining feature of pancreatic ductal adenocarcinoma (PDAC), the most common form of pancreatic cancer, and contributes to tumor progression and resistance to therapy. Pancreatic stellate cells (PSCs) are the main cell type causing the desmoplastic reaction. Quiescent PSCs in a normal pancreas act as lipid-storing cells with a limited secretome, whereas activated PSCs in the tumor microenvironment produce a vast array of secreted proteins implicated in cancer progression. Dr. Evans has hypothesized that restoration of the quiescent PSC phenotype will disrupt PSC–tumor cell communication and impair tumor growth. His lab has developed a screen to identify small molecules that revert PSCs to their quiescent phenotype using the accumulation of lipid droplets as a hallmark of the quiescent state.
ULK1 Inhibitors, in combination with mTOR inhibitors as a cytotoxic cancer therapy – Reuben Shaw Details»
The serine-threonine kinase mammalian target of rapamycin (mTOR) plays a major role in the regulation of cell growth, and metabolism. Given that the mTOR pathway is hyperactivated in a number of cancers, it has been proposed that mTOR inhibitors will have broad therapeutic application across many tumor types, and Rapamycin and its analogs (rapalogs) as well as ATP-competitive inhibitors of mTOR are being actively pursued as therapeutic candidates. In 2012, the National Cancer Institute listed more than 200 clinical trials testing the anticancer activity of these inhibitors, both as monotherapy and as a part of combination therapies for many cancer types. However, mTOR inhibitors as single agents have modest activity, and combination therapies are being developed with the idea of overcoming resistance to the mTOR inhibition, and increasing efficacy.
While the role of mTOR in cell growth is well known, its role in autophagy has only recently been identified. Autophagy is the cellular process by which cells break down intracellular proteins and organelles under conditions of stress, freeing up metabolic intermediates and promoting cell survival. Dr. Shaw’s work recently demonstrated that a complex controlling the initiation of autophagy, composed of a kinase called ULK1, is inactivated by mTOR (Egan et al., Science 2011). Thus, inhibiting mTOR will lead to activation of autophagy which is a major pathway promoting cell survival. The propensity of mTOR inhibitors to promote tumor cell survival via ULK1-dependent autophagy may help explain why rapalogs are primarily cytostatic, and only effective as disease stabilizers rather than for regression. Dr. Shaw has postulated that combining inhibition of ULK1 with rapamycin treatment might convert the standard cytostatic effect of mTOR inhibitors into a cytotoxic effect, once the survival benefit of ULK1-initated autophagy is removed. His laboratory has developed a variety of inducible, genetically-engineered mouse models that give biological readouts of treatment regimes in settings that are predictive of human cancer subtypes. He has also developed a vast array of in vitro assays to test biological hypotheses across all members of the relevant pathways, and to screen for ULK1 inhibitors. He is currently developing small molecule inhibitors with a collaborator.
New targets for triple negative breast cancer – Geoff Wahl Details»
Geoff Wahl’s work on the link between development and cancer has demonstrated that the invasive and proliferative processes of mammogenesis resemble phases of cancer progression. Expression profiles of isolated fetal mammary stem cells (fMaSC) and associated stroma exhibit striking similarities to profiles of triple negative and Her2+ breast cancer. Gene expression, transplantation and in vitro analyses revealed mechanisms, including ErbB and FGF pathways that regulate fMaSC growth. These and other candidates potentially provide new targets for diagnosis, prognosis and therapy of cancer.
Mouse models of non-small cell lung carcinoma – Reuben Shaw Details»
Shaw’s models are based off an inducible K-ras oncogene expressed in mouse epithelium once the mice inhale an adenovirus expressing cre-recombinase. His lab crossed the K-ras model to a ubiquitous promoter driving conditional luciferase expression only in cells that Cre has entered. In this fashion, they derive a signal from the tumor cells proportional to tumor mass which arises spontaneously in vivo in the accurate setting of the lung and from the accurate genetic lesions found in human non-small cell lung cancer (NSCLC). They combined the conditional K-ras luciferase mice with mice conditionally inactivated for the p53 tumor suppressor, AMPK, or the LKB1/STK11 tumor suppressor. Importantly, K-ras, p53, and LKB1 are the three most commonly mutated genes in human NSCLC, and the K-ras p53 (KP) mice and K-ras LKB1 mice (KL) develop consistent aggressive carcinomas within 8 weeks of inhaling cre-recombinase which allows the scientists to rigorously test therapeutics in real time in these models.
Miscellaneous
High throughtput screen for modulators of protein-protein interactions (PPI) – Geoff Wahl Details»Many myths exist around the difficulties of developing PPI antagonists. As a result, this approach is severely under-exploited, despite the fact that the extensive human interactome presents opportunities that dwarf current drug targets. Professor Wahl and his team have developed a platform cell-based assay in high-throughput format to screen for inhibitors and agonists of protein-protein interactions. This novel-screening platform combines an inducible protein expression system with the bi-molecular luciferase complementation (BiLC) assay, allowing for the interrogation of transient and dynamic PPIs, identification of novel intracellular protein interaction partners, and screening and characterization of PPI antagonists and agonists. The assay was initially developed to screen for inhibitors of p53-MDM2/MDMX in cancer, but is broadly applicable to many protein-protein interactions, including K-Ras Inhibitors.
Motor neuron dysfunction in ALS and spinal muscular atrophy (SMA) – Sam Pfaff Details»
A major challenge in studying SMA has been the lack of appropriate model systems. Dr. Pfaff has developed a novel embryonic-stem cell-based model of SMA that phenocopies the pathology of human SMA, and can be used to study the basis of the disease and screen for compounds that might increase survival of motor neurons. Through these studies he has identified a new transcription factor and associated pathway that could yield new avenues for therapeutic intervention in SMA.

